Alternative names for phaeochromocytoma
Phaeo; Pheochromocytomas; Chromaffin tumours; Intra-adrenal (i.e. that grows within the adrenal gland); paragangliomas
Phaeochromocytomas and paragangliomas are closely related tumours. The main difference between them is where they grow. A tumour that forms inside the adrenal gland is called a phaeochromocytoma. A tumour that grows outside the adrenal gland, often along the nerves in the neck, chest, abdomen, or pelvis, is called a paraganglioma. They often behave similarly and involve the same hormones and genetic causes.
The term phaeochromocytoma is still used for adrenal tumours, but many doctors now group both types under the same umbrella term for phaeochromocytomas and paragangliomas (PPGL), especially when discussing genetic testing, management, and long-term follow-up.
What is a phaeochromocytoma?
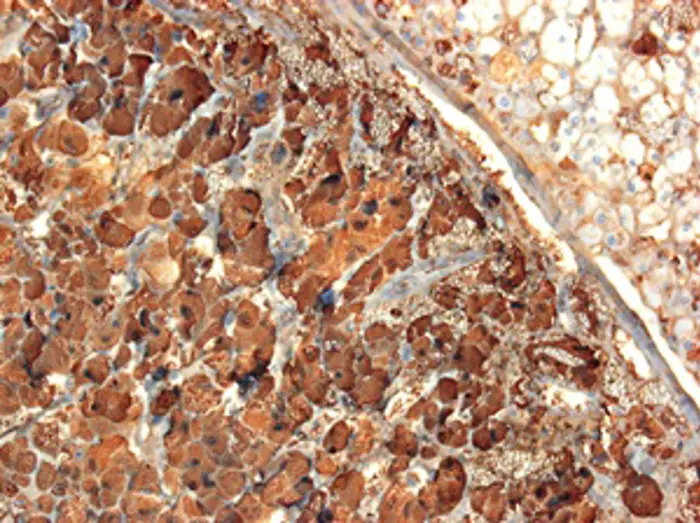
Figure 1: Light micrograph of a section through a phaeochromocytoma tumour of the adrenal gland. At left in the medulla, large tumour cells are seen arranged in cords or small masses marked by chromogranin A (blue); at upper right the normal adrenal cortex is seen.
A phaeochromocytoma is a rare tumour that forms in the adrenal medulla, the central part of the adrenal gland. Humans have two adrenal glands, each sitting on top of each kidney. The adrenal medulla is responsible for the production of hormones, known as catecholamines, such as:
- Adrenaline (epinephrine)
- Noradrenaline (norepinephrine) and
- Dopamine (in smaller amounts)
These hormones are needed for the ‘fight or flight response’ when the body becomes ready for stressful events or increased physical activity.
A phaeochromocytoma causes the adrenal gland to make an excess (too much) of these hormones, which can affect blood pressure, heart rate, and other body functions.
Phaeochromocytomas are related to a similar type of tumour found outside the adrenal gland called a paraganglioma.
What causes phaeochromocytomas?
In many cases, the cause of phaeochromocytomas is not known. However, up to 40% of phaeochromocytomas and paragangliomas (PPGLs) are due to an inherited (genetic) cause. Today, more than 20 genes are known to increase the risk, and about half of the inherited cases are due to mutations in the succinate dehydrogenase (SDH) genes.
What are the signs and symptoms of a phaeochromocytoma?

Figure 2: Symptoms of phaeochromocytomas. Image created using Biorender.
Phaeochromocytomas may not cause any symptoms and may only be found by chance during a scan for something else.
However, signs and symptoms can be caused by either excess hormone being released into the bloodstream, local growth or the distant spread of a tumour.
The release of high levels of hormones like adrenaline and noradrenaline can cause very high blood pressure (can be up and down or can stay high). Other symptoms include headaches, a fast or pounding heartbeat, sweating and feeling anxious or panicky. Patients may describe a ‘feeling of impending doom’.
These hormones may be released in bursts, so symptoms may come and go and can often be mistaken for panic attacks.
Signs and symptoms may also relate to the tumour growing and pressing on other nearby organs. This can cause pain or discomfort in the abdomen or back.
If the tumour remains undiagnosed and hormone levels are very high for a long time, patients can have damage to their heart and kidneys. Occasionally patients can also become very ill and the disease may become life-threatening.
How common are phaeochromocytomas?
Phaeochromocytomas and paragangliomas (PPGLs) are rare tumours. An estimated two to eight new cases occur per million people each year. These tumours are found in up to one in ten of adrenal growths that are picked up during scans (Wolf et al 2024, National Cancer Institute, 2024).
They also cause high blood pressure in up to six in every 1000 patients with persistent high blood pressure (Jonsson et al, 2023).
Most phaeochromocytomas are non-cancerous (benign), although around one in ten are cancerous (malignant) and may spread to other parts of the body.
Are phaeochromocytomas inherited?
Phaeochromocytomas may occur randomly (sporadic) or be inherited in families (familial). They occur most commonly in middle age (30-60 years) but can also occur in children.
Approximately one in four people with a phaeochromocytoma have a familial (inherited) form caused by a genetic abnormality (mutation) (Newcastle NHS Trust, 2022). Inherited phaeochromocytomas may occur as part of genetic syndromes, which include:
- Von Hippel-Lindau disease
- Multiple endocrine neoplasia type 2a
- Multiple endocrine neoplasia type 2b
- Familial paraganglioma syndromes
Having a genetic mutation does not mean that all members of the family will develop a tumour, but it may increase the risk. Some genetic abnormalities may be associated with other types of tumours.
How is a phaeochromocytoma diagnosed?
If a patient does not have symptoms, phaeochromocytomas may remain undiagnosed. They are typically found by chance when having an abdominal scan for an unrelated reason.
If the patient has symptoms, the doctor may carry out an examination including an assessment of the blood pressure and fast or irregular pulse. Initial tests may include:
Collection of urine over 24 hours or a blood test to check for high levels of the hormones adrenaline and noradrenaline, or their by-products.
If those levels are raised, specialised imaging is carried out which includes:
- Magnetic resonance imaging scan (MRI)
- Computerised tomography (CT) scan
- Nuclear medicine scans including metaiodobenzylguanidine (MIBG) or a positron emission tomography (PET) scan to confirm the site of the tumour and check whether there is any evidence of tumour spread.
All these tests usually occur on an outpatient basis (i.e. admission to the hospital is not required). If a genetic cause is suspected, blood samples may be taken to carry out genetic testing.
How is a phaeochromocytoma treated?
Treatment usually begins with medications to block the effects of excess hormones and stabilise blood pressure before surgery. These medications are called alpha-blockers (phenoxybenzamine, doxazosin, prazosin, terazosin) and beta-blockers (propranolol, labetalol). Beta-blockers should only be started after alpha-blockers, to avoid sudden dangerous rises in blood pressure.
Surgical removal of the tumour is the best curative treatment. Surgery should be carried out by a surgeon and anaesthetist experienced in operating on this type of tumour and in centres where there is a multidisciplinary endocrine team that deals with these tumours.
If the tumour cannot be fully removed, surgery is not possible or has spread to other parts of the body, then other treatments include chemotherapy and/or a radioactive form of treatment called radiolabelled MIBG therapy. Newer targeted drug treatments exist, depending on tumour type and genetic background (e.g. a genetic mutation known as SDHB mutation). An example is a medication known as a tyrosine kinase inhibitor (e.g., sunitinib).
Another form of treatment for some advanced tumours is peptide receptor radionuclide therapy (PRRT), such as 177Lu-DOTATE. This uses a small molecule that targets the tumour cells and delivers radiation directly to them, helping to slow or shrink the tumour.
Are there any side-effects to the treatment?
Medications that block hormones like adrenaline and noradrenaline may cause side-effects including dizziness on standing up, low blood pressure, blocked nose and cold hands and feet.
Risks of surgery include bleeding, infection or damage to other nearby organs, e.g. kidney or the spleen. These risks are low when surgery is performed by a specialist endocrine team.
What are the longer-term implications of a phaeochromocytoma?
Many patients with a phaeochromocytoma return to very good health after treatment. If the tumour is completely removed at initial surgery, most patients are cured. However, patients need long-term follow-up to make sure that no new tumour develops in the future. This is unusual but happens in about one in ten of patients with phaeochromocytoma.
Follow-up care includes blood pressure monitoring and occasional measurement of adrenaline and noradrenaline related chemicals in urine or blood. Having a genetic mutation may increase the risk of a further tumour and further scans might be needed in the following years for monitoring.
If the tumour is linked to a genetic condition, other family members may need genetic counselling or testing.
If the phaeochromocytoma cannot be fully removed or it spreads, it usually grows slowly, and many patients may still live for many years with the help of ongoing treatment.